Oncohistones; Epigenetic drivers of Cancer and Beyond
by Wajih Jawhar
Genetic mutations impacting epigenetic processes are highly recurrent and can be pathognomonic in cancer. One class of such mutations directly alters the histones themselves, creating “oncohistones”. These oncohistones drive transformation by disrupting the genome-wide landscape of post-translational modifications (PTMs) on histone residues. Functional analysis of oncohistones has served to delineate disease mechanisms and expanded our understanding and appreciation of epigenetic control of cellular processes.

H3K27M:
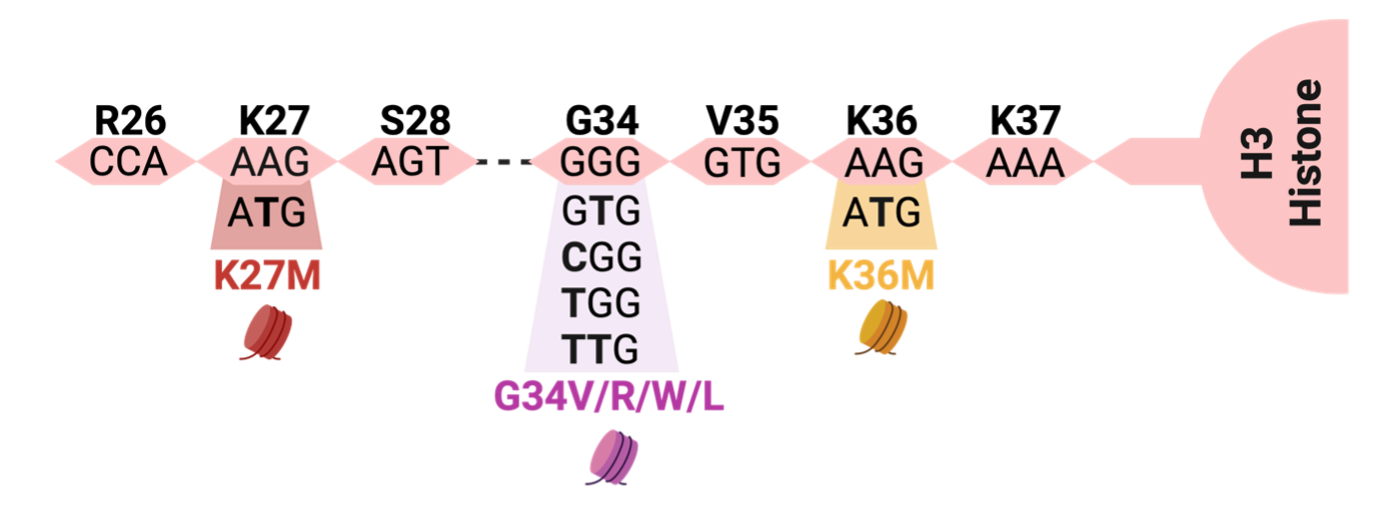
One of the most extensively studied oncohistones is H3K27M, where the 27th amino acid residue of the Histone H3 tail is changed from lysine (K) to methionine (M) (Fig. 1). H3K27M can occur in any of the histone H3 variants: canonical H3.1/H3.2 or noncanonical H3.3. H3K27M reshapes the chromatin landscape by inhibiting the activity of Polycomb Repressive Complex 2 (PRC2), a key protein complex responsible for depositing mono-, di-, and tri-methyl marks on H3K27. By depositing these repressive marks, PRC2 orchestrates normal development in space and time by influencing the expression of key developmental genes in stem cells that contribute to the formation of the ~300 differentiated cell types that comprise the human body (Harutyunyan et al., 2019; Yu et al., 2019).
H3K27M oncohistones were initially discovered and characterized in aggressive pediatric brain tumors in the brain midline, collectively termed “diffuse midline gliomas, K27-altered”. Growing interest and extensive investigation of these oncohistones led to their subsequent discovery in other cancers of the central nervous system (CNS) (PFA ependymomas) and more rarely in subsets of white blood cell cancer (acute myeloid leukemia) and bone cancers (Osteosarcoma) (Krug et al., 2021; Salomoni et al., 2023).
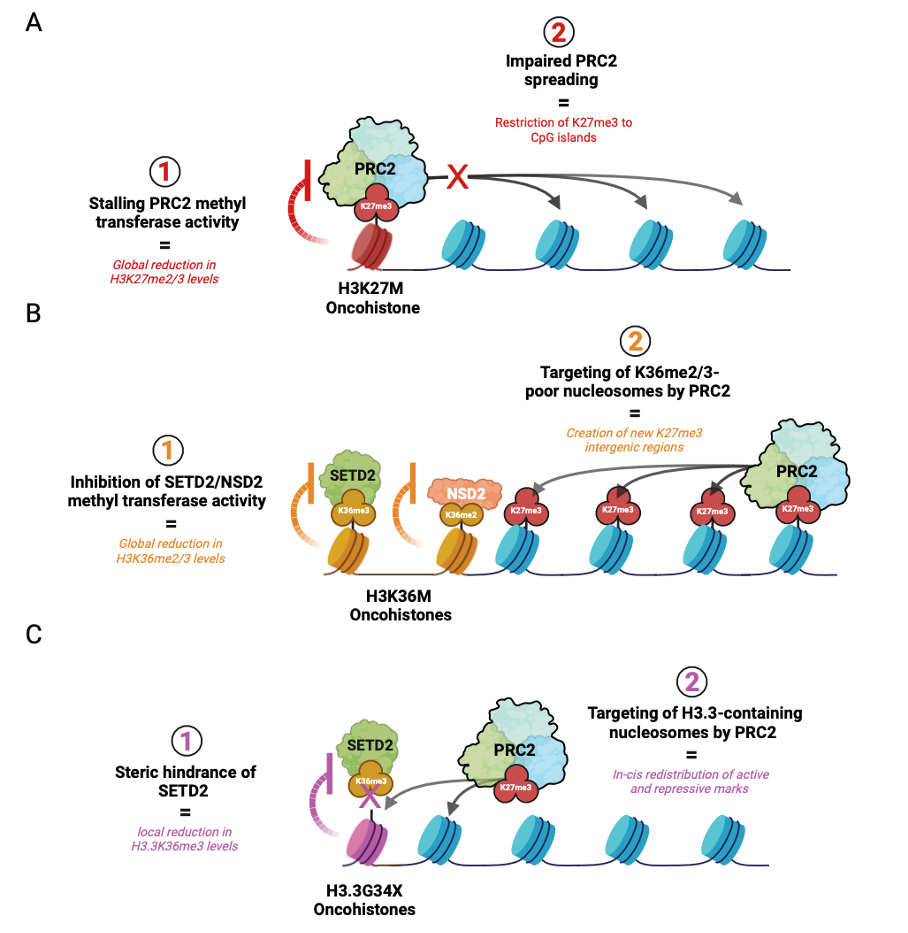
Throughout development, PRC2 is initially recruited with the help of accessory proteins (e.g. MTF2 and JARID2) to deposit H3K27me3 at CpG-dense regions called CpG islands. This process is referred to as “nucleation”, signifying the initial establishment of H3K27me3 foci on chromatin. PRC2 is then allosterically activated by binding its H3K27me3 product to induce rapid “spreading” of H3K27me2 and me3 onto adjacent chromatin and distally through long-range interactions of nucleation hubs (Oksuz et. al, 2018). However, in cells that acquire H3K27M, PRC2 stalling results in a global decrease in H3K27 di- and tri-methylation and its impaired spreading from CpG islands. Given that H3K27me3 catalysis is enzymatically more demanding than H3K27me2, these effects are more prominent on the former histone mark (Fig. 2A). Functionally, this phenomenon creates an epigenomic landscape that reinforces a proliferative stem-like state associated with transformation (Harutyunyan et al., 2019; Boileau et al., 2019).
H3K36M
Similar to Lysine 27, the neighboring Lysine 36 (K36) residue is also a mutational hotspot for K-to-M mutations in specific cancer subtypes. Most of our knowledge on K36M oncohistones comes from mesenchymal cancers such as chondroblastoma, chondrosarcoma and undifferentiated pleomorphic sarcoma that harbor H3.1/H3.3K36M mutations (Lu et al., 2016; Behjati et al., 2013). Albeit K36M was initially discovered in a subset of head and neck squamous cell carcinomas. (Schapira et al., 2014) and more recent work dissects its role in immune evasion in these cancers (Li et al., 2022).
H3K36M is a potent inhibitor of NSD2 and SETD2 proteins that are responsible for depositing H3K36me2 and me3 histone marks, respectively. The former PTM exhibits broad distribution across the genome and demarcates intergenic regions, while the latter is enriched in actively transcribed gene bodies. In addition to promoting gene expression, K36 methylation antagonizes PRC2-mediated silencing by disrupting the interaction between the H3 histone tail and the active site of EZH2, the catalytic subunit of PRC2 (Lu et al., 2016, Fang et al., 2016; Muller et al., 2020). Given the functional antagonism between K36 and K27 methylation, genome-wide reduction of H3K36me2/3 creates a new histone environment that becomes more permissive to PRC2 targeting.
In a study that investigated this inverse relationship in the context of K36M sarcomas, K36M expression did not result in elevated genic H3K27me3 levels but created new H3K27me3 intergenic domains in regions that lost H3K36me2 (Fig. 2B) (Lu et al., 2016). Consequently, the redistribution or “dilution” of genic H3K27me3 led to a corresponding dilution of canonical polycomb repressive complex 1 (cPRC1), a reader of H3K27me3. Upon recruitment of cPRC1 to H3K27me3-demarcated polycomb targets, cPRC1 monoubiquitinate lysine 119 on histone H2A (H2A119ub) to promote heterochromatin formation and gene repression (Gao et. al, 2013). In H3K36M-expressing mesenchymal progenitors, the dilution of cPRC1 from these targets permit aberrant de-repression of genes that impede stem cell differentiation and promote sarcomagenesis. (Lu et al., 2016, Fang et al., 2016).
H3G34X
A common feature of K36M and K27M is their occurrence on lysine residues that undergo PTMs. However, not all 59 amino acid residues on the H3 tail undergo these modifications. Interestingly, mutations in non-chemically modified residues such as Glycine 34 (G34) are also implicated in diseases such as cancer.
Glycine 34, located 2 residues away from Lysine 36, can harbor 4 different oncohistone mutations; H3 G34W, G34L, G34R, or G34V that change Glycine to Tryptophan, Leucine, Arginine, or Valine respectively (Fig. 1). The first 2 amino acid changes (G34W/L) most commonly occur in connective tissue (mesenchymal) tumors, while the latter 2 (G34R/V) commonly occur in tumors of the brain cortex named “diffuse hemispheric gliomas, G34-altered”. (Behjati et al., 2013; Khazaei et al., 2020)
Unlike H3K27M and H3K36M that change the behavior of enzymes chemically, referred to as allosteric inhibition or modulation, G34X oncohistones function through physical interference. This occurs because glycine, a small amino acid, is changed to a bulky residue such as tryptophan, creating steric hindrance. Additionally, unlike H3K27M and H3K36M, G34X mutations exclusively occur on H3.3 histones, potentially indicating a key requirement of its location on chromatin for pathogenesis. As the enzyme SETD2 is recruited to tri-methylate K36, the now-bulky residue physically blocks this reaction. This phenomenon is known as “steric inhibition” or “steric hindrance” relating to the spatial/physical arrangement of a molecule. This causes a local loss of K36 tri-methylation at actively transcribed genes leading to aberrant K27 methylation by PRC2, resulting in silencing of these loci (Fig. 2C).
In the giant cell tumors of the bone (GCTB), H3.3G34W renders transcriptional changes that drive abnormal bone resorption (bone breakdown) and local invasion of surrounding bone tissue, a characteristic feature of this tumor entity. (Jain et al., 2020; Khazaei et al., 2020). Further studies on G34 oncohistones continue to unravel their function in various diseases such as neurodegeneration and their disease mechanisms that bridge aberrant histone methylation with DNA methylation patterns (Khazaei et al., 2023).
In all, this intricate balance between histone modifications, collectively known as the histone code (Allis and strahl, 2000) remains an active area of investigation. Gene expression is not solely controlled by the presence or absence of a specific histone mark, but also by its relative concentration and dilution on chromatin and its interaction with the rest of the epigenetic toolbox.
Acknowledgments
We would like to extend our sincere thanks to Dr. Martin Hirst for his expert review, which greatly strengthened this article. Special appreciation goes to Reem El Kabbout for providing the French translation, making these articles accessible to a broader audience and Dr. Shaghayegh Nouruzi for her editing.